Before we dig in, I want to emphasize that this is science, not tribal warfare. The goal is to arrive at the best answer, rather than to win an argument. I’m proceeding in good faith, based on my belief that Ludwig and I are both serious people who care about science and human health, and I hope my audience will do the same. That said, let’s get to it.
Introduction: CICO vs. insulin. Or is there a third model?
Similar to Gary Taubes, Ludwig presents a choice between the calories-in, calories-out model (CICO) of obesity, and the insulin model. The CICO model is the idea that our body weight is determined by voluntary decisions about how much we eat and move, and in order to control our body weight, all we need is a little advice about how many calories to eat and burn, and a little willpower. The primary defining feature of this model is that it assumes that food intake and body fatness are not regulated. This model seems to exist mostly to make lean people feel smug, since it attributes their leanness entirely to wise voluntary decisions and a strong character.
I think at this point, few people in the research world believe the CICO model. Ludwig and I both agree that it provides a poor fit for the evidence. As an alternative, Ludwig proposes the insulin model, which states that the primary cause of obesity is excessive insulin action on fat cells, which in turn is caused principally by rapidly-digesting carbohydrate. According to this model, too much insulin reduces blood levels of glucose and fatty acids (the two primary circulating metabolic fuels), simultaneously leading to hunger, fatigue, and fat gain. Overeating is caused by a kind of “internal starvation”. There are other versions of the insulin model, but this is the one advocated by Ludwig (and Taubes), so it will be my focus.
But there’s a third model, not mentioned by Ludwig or Taubes, which is the one that predominates in my field. It acknowledges the fact that body weight is regulated, but the regulation happens in the brain, in response to signals from the body that indicate its energy status. Chief among these signals is the hormone leptin, but many others play a role (insulin, ghrelin, glucagon, CCK, GLP-1, glucose, amino acids, etc.).
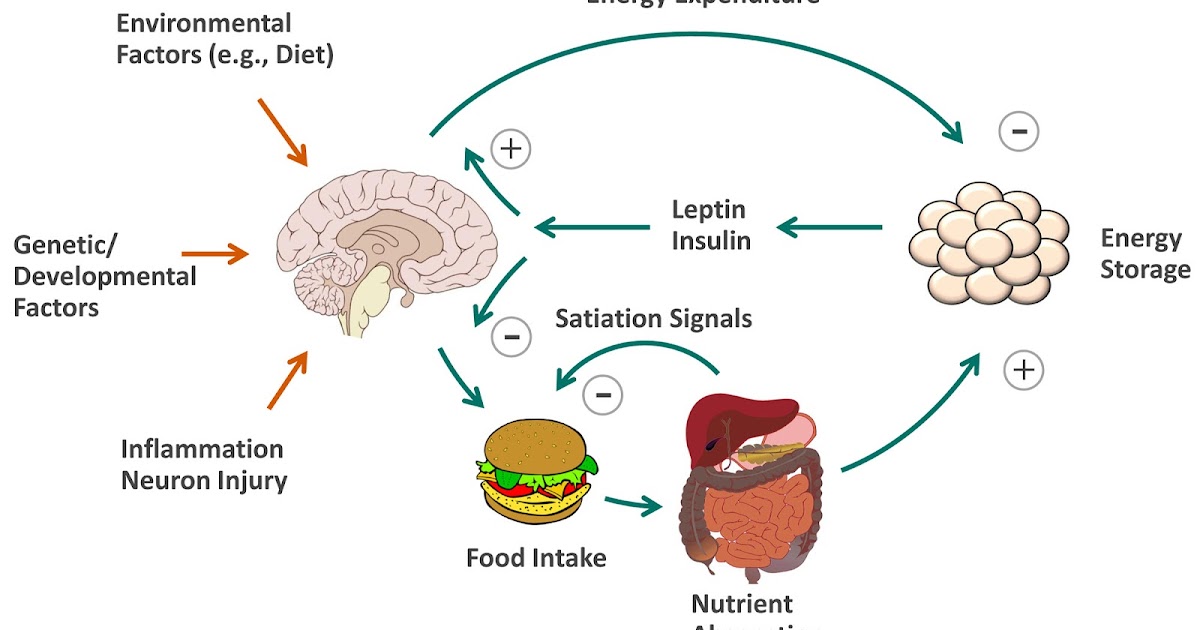
Here is a simplified schematic overview of how the system works, from a 2012 review paper I wrote with my scientific mentor Mike Schwartz, titled “Regulation of food intake, energy balance, and body fat mass” (1). This figure summarizes more than a century of research in our field:
Here’s the gist of it: there are negative feedback loops between the brain and fat tissue, and between the brain and the gut. These are what regulate body fatness and appetite. The primary known feedback signal that regulates body fatness is leptin– a fact that has remained scientifically unchallenged since shortly after its identification in 1994. Insulin plays a role as well, acting directly on the brain in a manner similar to leptin, although much less powerfully. As you can see, this model doesn’t resemble the CICO model– or the insulin model.
Regulation happens principally as a result of the brain changing the number of calories entering and leaving the body (in humans, mostly entering)– so the much-maligned calorie maintains a central role in the process. Even though calories aren’t the first link in the causal chain, they are nevertheless a critical link.
Most people in my field also believe that calorie intake is determined both by hunger (homeostatic eating), and factors other than hunger (non-homeostatic eating). I agree with them.
So this third model acknowledges the importance of regulation, the importance of unregulated factors, and the importance of calories. Although it’s more complex than the insulin and CICO models, it has the advantage of being supported by a large body of evidence. Let’s call it the leptin model, for simplicity’s sake. I won’t spend much more time on it because it’s peripheral to this debate, but I did want to articulate it as a third model so we aren’t limiting our choices to 1) a CICO model that is obviously wrong, and 2) the insulin model.
I’m not going to do a point-by-point response to Ludwig’s point-by-point response, because that would be exceedingly cumbersome. What I’ll do instead is try to distill out the most important concepts, and discuss those. In particular, I’ll be focusing on key lines of evidence that have the potential to support– or falsify– the insulin model. This is where the rubber meets the road for hypothesis testing.
Is obesity caused by internal starvation?
In my initial post, I pointed out that people with obesity have normal or elevated levels of blood glucose and fatty acids– not lower levels as the insulin model predicts (1a, 1b, 1c). This suggests that 1) they are not suffering from “internal starvation”, 2) they are not suffering from excessive insulin action (including on fat cells), and 3) therefore this mechanism cannot explain the well-established fact that people with obesity tend to eat about 20 percent more calories than lean people (2, 3).
Ludwig countered that “static analysis after obesity has developed, rather than during its dynamic stages, are misleading”. In other words, I was focused on the wrong phase of obesity; we should be looking at the weight gain phase instead. Let’s think through the implications of this statement. It implies that internal starvation makes us overeat as we’re gaining weight, but doesn’t explain continued overeating once we reach the obese state, or the fact that the obese state persists after internal starvation is no longer occurring. It therefore also requires the existence of two separate mechanisms that cause overeating, one involving internal starvation and one not, and requires a hand-off between them at some point in time. I’m not aware of experimental evidence that supports any of this.
Let’s take a closer look at the weight gain phase, as suggested. Is there evidence that people who are gaining weight have lower glucose and fatty acid levels than people who aren’t? There is not. I’m not aware of any evidence in humans or in animal models of diet-induced obesity that circulating glucose or fatty acid levels are lower during active weight gain. In Mike Schwartz’s lab, we have done countless glucose measurements in mice and rats gaining weight rapidly due to a fattening diet, and we have also measured circulating fatty acid levels at times, and neither one is decreased during active fat gain (3a).
Ludwig acknowledges this lack of evidence in his article, but instead focuses on indirect evidence that’s not particularly relevant to common obesity, such as hypothalamic lesions in rodents (As an aside, Bruce King has shown that the fattening effect of VMN lesions does not depend on increased insulin levels (4), and John Brobeck showed in the 1940s that these lesions cause voracious overeating as soon as the anesthesia wears off, not after some period of altered fat tissue metabolism (5). In any case, alterations in fat tissue metabolism are the result of the brain lesion, showing that the brain is influencing fat tissue metabolism in these experiments, not the reverse).
So in summary, there is no direct evidence that internal starvation occurs during 1) weight gain or 2) the overweight/obese state, and there is some direct evidence that it does not occur. The internal starvation hypothesis rests entirely on indirect evidence that I don’t find particularly pertinent.
But let’s take a step back for a minute. Are low circulating fatty acid levels able to activate the brain’s hunger circuits in the first place, leading to increased food intake and weight gain? Because if they aren’t, then it’s hard to understand how insulin (or anything else) would lead to overeating by reducing fatty acid levels. This is testable. To test it, we would want to reduce circulating fatty acid levels and see if it provokes a hunger response, and ideally, weight gain. Fortunately for us, this study has been done.
Hideo Makimura and colleagues recently published the results of a 6-month randomized, placebo-controlled study in which they used a drug called acipimox to chronically reduce circulating free fatty acid levels in people with obesity (6). The drug caused a substantial (38%) reduction of free fatty acid levels for the duration of the 6-month study.
According the idea of internal starvation, the acipimox group should have been ravenously hungry and gained fat rapidly, and energy expenditure should have declined as well. Yet the researchers reported that “caloric and relative macronutrient intake did not change significantly between groups”. Neither group’s BMI or body composition changed significantly, nor did their energy expenditure. The study was published in the Journal of Clinical Endocrinology and Metabolism, a respected journal in my field.
The brain monitors a number of signals from the body to measure energy status, and it uses these to set your appetite. Apparently, low circulating fatty acid levels are not one of the signals the human brain pays attention to when it sets appetite. This seriously undermines the concept of internal starvation, and consequently the insulin model.
In contrast, we have strong evidence that the human brain pays close attention to circulating levels of leptin. When leptin levels are low, whether due to weight loss or mutations in the leptin gene, it triggers a full-blown starvation response including increased hunger, increased responsiveness to food cues, and reduced energy expenditure. Bringing leptin levels back to normal via injection terminates this starvation response, clearly demonstrating that low leptin was the principal cause (7, 8, 9, 10, 11, 12, 13). These results don’t leave much room for insulin or fat cell metabolism to be involved in the body’s starvation response (except perhaps as downstream effectors of leptin).
Do high insulin levels predict fat gain?
This is a straightforward prediction of the insulin model, and we have many human studies with which to test it. In my initial post, I cited a systematic review paper that summarized the results of 22 prospective studies examining this prediction (14). Overall, the literature suggests that people with high insulin levels do not gain more weight or fat over time than people with low insulin levels.
Ludwig countered by arguing that what matters isn’t insulin levels, but insulin action; in other words, how much of an impact the insulin actually has on cells and tissues. Insulin action is determined both by insulin levels and insulin sensitivity (how well tissues “hear” the insulin signal). This is the same argument I have used to question the insulin model, because proponents often assume that high insulin levels automatically imply elevated insulin action. They generally overlook the fact that people with high insulin are almost invariably insulin resistant (including their fat tissue), and that insulin action is normal or reduced (as judged by normal or elevated blood glucose and fatty acids, and normal or elevated whole-body lipolysis rates).
In fact, this is precisely the reason why insulin injections, insulin-producing tumors, and similar non-physiological examples cited by Ludwig and Taubes are irrelevant to the question at hand. These arguments attempt to draw an analogy between non-physiological insulin excess and garden-variety hyperinsulinemia– but in the latter case, there is no evidence that insulin action on fat cells is actually increased, and a fair bit of evidence that it isn’t.
In any case, I agree with Ludwig’s basic premise that insulin action is what really matters. Yet if we return to the review paper I cited, it didn’t just consider studies that measured fasting insulin levels. The studies measured multiple insulin-related variables, including fasting insulin levels, insulin sensitivity, and the insulin response to a glucose challenge. Although we could cite individual studies to support any hypothesis we want, the overall literature suggests that none of these variables are reliably associated with weight gain. In fact, I’m not aware of any insulin-related variable that is reliably associated with weight or fat gain in humans, despite intensive research in this area. Perhaps we’re not measuring the right thing, or not measuring in the right way, but that possibility is nothing more than speculation at this point.
Ludwig suggests that we should be looking for the effects of insulin during the weight gain phase. That is exactly what these studies have done, and they did not find that weight gain was consistently associated with a distinct insulin signaling profile in any measured way.
So we’ve measured insulin levels, insulin sensitivity, and the insulin response to carbohydrate. We’ve measured them in people who are gaining weight and people who aren’t. We’ve measured them in multiple races and ages. And no clear pattern has emerged suggesting that insulin signaling might be playing an important role in the fattening process. Again, it’s always possible that we haven’t measured the right variable yet, or haven’t measured it in the right way, but that is nothing more than speculation at this point. Currently I find this evidence rather difficult to reconcile with the insulin model.
Do low-glycemic diets work for fat loss?
If rapidly-digesting carbohydrate that markedly elevates insulin levels is a major driver of overeating and weight/fat gain, then low-glycemic diets that reduce insulin exposure should be an effective tool for reducing food intake and body weight. In my initial post, I reviewed evidence suggesting that 1) the glycemic index doesn’t reliably predict the satiety response to common foods, and 2) low-glycemic diets are ineffective for weight control.
To the first point, Ludwig countered that the study I cited only measured the satiety response over a two-hour period, but hunger usually occurs later (15). He also cited a review paper he wrote in 2002 suggesting that single-meal studies have generally found that low-glycemic meals are more sating than high-glycemic meals (16). This is a fair point, and the cited evidence does support it (although many of these studies were not controlled for other differences known to affect satiety, such as fiber content, palatability, and calorie density). However, findings in this area haven’t been especially consistent, and in a few cases high-glycemic foods were actually more sating (17, 18, 19). I do acknowledge that the weight of the evidence is in his camp on this point.
But in any case, what we really care about is not what happens at a single meal, but what happens over the long haul. That is the question my second point addresses. We have quite a bit of evidence suggesting, fairly consistently, that low-glycemic diets don’t work for weight loss (20, 21, 22, 23, 24, 25, 26).
Ludwig countered that these studies “suffer from severe non-compliance, limiting inferences”. In other words, people don’t follow the experimental diet very faithfully, explaining why the diets appear ineffective. While I agree that adherence is always a limiting factor in human trials that don’t lock people up in a research ward, poor adherence doesn’t explain the null results in this case.
For one thing, adherence is always a problem in human diet trials, but they nevertheless often report weight loss. This has been shown for the low-carb diet, the Paleo diet, the vegan diet, the Mediterranean diet, simple portion control, and even the embattled low-fat diet. Some of these are tough diets that people have a hard time sticking with, but they still cause measurable weight loss despite imperfect adherence. In diet trials, adherence rates to low-glycemic diets are similar to those of other diets, so it’s hard to understand how this would explain the lack of efficacy.
But let’s get specific. I’d like to start with a study that may be the purest test of the hypothesis available (27). It was conducted by Walter Willett and Frank Hu, two of Ludwig’s colleagues at Harvard.
They randomized 203 healthy women (average BMI = 27) to one of two diets: high-glycemic or low-glycemic. Differences in glycemic index were achieved primarily by eating different types of rice that have different digestion speeds, so the study was relatively well controlled for other aspects of diet like fiber, calorie density, palatability, etc. The overall glycemic index (and glycemic load) of the diets differed by two-fold (40 vs. 79), which implies a large difference in both glucose and insulin exposure. This large difference was sustained for the full 18 months of the intervention.
There were no significant differences in hunger or calorie intake between the two groups. At two months, there was a trivial difference in body weight of 0.4 kg favoring the low-glycemic group, but this disappeared by the end of the study. The conclusion is that large and sustained differences in post-meal glucose and insulin exposure have no meaningful long-term impact on hunger, food intake, or body weight in women (who were overweight on average).
Let’s consider another study– the one from Ludwig’s group that I cited previously (28). This 18-month randomized trial pitted a low-glycemic-load diet against a low-fat diet in 73 obese young adults. The low-glycemic diet was focused on whole foods like vegetables, beans, and fruit, at the expense of refined grains, starchy vegetables, fruit juice, and sweets. The low-fat diet was focused on low-fat grains, vegetables, fruits, and beans, at the expense of of added fats, sweets, and high-fat snacks. The low-glycemic diet was a bit lower in carbohydrate (40% vs 55%), and both diets were fairly high in protein (25%). Neither diet was calorie restricted.
Glycemic load differed substantially on the two diets, and a difference was maintained over the 18-month study, although it narrowed at 12 and 18 months. This implies substantial differences in glucose and insulin exposure.
So what happened? Hunger, calorie intake, and participant satisfaction didn’t differ between groups. Both groups lost weight (4-5 kg at max), but the magnitude and trajectory of weight loss between the two groups was virtually identical (see graph below). Substantial differences in diet-related glucose and insulin exposure did not result in detectable differences in hunger, calorie intake, weight loss, or weight regain.
 |
| 18-month weight changes in Ebbeling et al. Note the p-value of 0.99, indicating that the two trajectories are statistically indistinguishable. |
In his response to my post, Ludwig pointed out that subjects who had a greater insulin response to a glucose challenge at baseline experienced more weight loss on the low-glycemic-load diet. This is true, but we have to keep the finding in context. The study wasn’t randomized to answer this question (unlike a recent study from Christopher Gardner’s group; 29), so this is what’s called an “exploratory analysis”. These carry less weight than the primary outcomes of a study, which are those that I discussed above. Exploratory analyses can certainly be informative, and there’s nothing wrong with conducting and publishing them, but they are better suited for generating hypotheses than for testing hypotheses. And they never trump the primary outcomes of a study.
The most straightforward interpretation of the study is that if you’re a young adult with obesity, reducing the glycemic load of your diet, and consequently your insulin exposure, has no special ability to curtail your appetite or help you lose weight. You might as well go on a low-fat, high-carbohydrate, high-glycemic diet. This is consistent with the results of the previous study I discussed, and the rest of the literature as a whole.
It remains possible that low-glycemic diets are helpful for a subset of people, but current evidence suggests they are not very effective as a general strategy for managing appetite and weight– counter to what the insulin model predicts.
Conclusion
In summary, several key predictions of the insulin model are not supported by the evidence, explaining why this model doesn’t get much traction in my field. There is essentially no direct evidence that the proposed mechanism occurs during or after normal weight gain, a fair amount of direct evidence that it doesn’t, and the arguments in favor of it are based on indirect evidence whose relevance to common obesity is often questionable.
I thank Dr. Ludwig for the opportunity to clarify my thoughts on this, and I wish him luck in his ongoing research.


{kind=link}